Dunham Lab Chemostat Manual
Maitreya Dunham
Last revised December 2005
This comprehensive manual covers the entire process of running a chemostat, including media recipes, chemostat setup, inoculation, data acquisition and storage, daily monitoring, harvests for DNA and RNA, and data analysis. Although I wrote it with my own yeast cultures and ATR Sixfors setup in mind, many of the procedures are general. If you have edits or additions to the manual, please email me at maitreya@princeton.edu
Please feel free to point other people to these instructions. Also, I would appreciate the citation if you use any of this information in a publication or talk.
Visit genomics.princeton.edu/dunham for the most recent updates to the protocol and for my other protocols on microarrays.
Thanks to Matt Brauer, my long-time companion in the chemostat lab, for help developing these protocols and for proofing the manual. Also thanks to Frank Rosenzweig who taught me how to run my first glass-blown chemostats. Many of the protocols were influenced by his chemostat aesthetic.
Edits since last version: new phosphate limitation recipe, new DNA prep, new filter-sterilization method of media preparation
© Maitreya Dunham 2005
Contents
Contents................................................................................................ 2
Figures................................................................................................... 5
Planning the experiment.................................................................. 6
Signup....................................................................................................... 6
Strains....................................................................................................... 6
Limitations................................................................................................. 7
Dilution Rate.............................................................................................. 9
Chemostat media............................................................................... 11
Salts........................................................................................................ 11
10X salts for carbon,
leucine, or uracil limitation................................... 12
10X salts for phosphate
limitation........................................................ 12
10X salts for sulfur
limitation................................................................ 12
10X salts for nitrogen
limitation............................................................ 12
Metals...................................................................................................... 13
1000X metals........................................................................................ 13
Vitamins................................................................................................... 14
1000X Vitamins..................................................................................... 14
Tubing and Fittings.................................................................................. 15
Carboy..................................................................................................... 15
Autoclaving the salts............................................................................... 16
Additives.................................................................................................. 18
glucose (dextrose)
limitation additives................................................. 19
phosphate limitation
additives.............................................................. 19
sulfur limitation
additives...................................................................... 19
nitrogen limitation
additives................................................................. 20
leucine limitation
additives.................................................................... 20
uracil limitation
additives...................................................................... 20
Making a carboy of
media........................................................................ 21
Attaching the carboy................................................................................ 21
Plates...................................................................................................... 23
YPD....................................................................................................... 23
YNB....................................................................................................... 24
Supplements for YNB............................................................................ 25
canavanine plates................................................................................ 26
Dropout powder................................................................................... 26
5-fluorouracil plates.............................................................................. 27
alpha-aminoadipate
plates................................................................... 27
Kanamycin (aka G418 or
Geneticin) plates........................................... 27
Assembling the vessel............................................................................. 28
Autoclaving the
chemostats..................................................................... 31
Setting up a run................................................................................ 32
Polarizing the
dissolved oxygen probes.................................................. 32
Setting up the vessel............................................................................... 32
Connecting the air................................................................................... 33
Starting the media flow........................................................................... 33
Programming the
chemostat.................................................................... 35
Profiles.................................................................................................. 35
Calibrating the
Dissolved Oxygen Probe.................................................. 39
Calibrating the
temperature probe.......................................................... 40
Inoculating the
chemostats..................................................................... 40
Signup..................................................................................................... 42
Data collection......................................................................................... 43
Sample tracking....................................................................................... 45
Preparing to sample................................................................................ 45
Contamination issues.............................................................................. 46
Sampling.................................................................................................. 46
Klett...................................................................................................... 46
Glycerol stock........................................................................................ 47
Spectrophotometer............................................................................... 47
Sonicator.............................................................................................. 47
Coulter counter..................................................................................... 48
Plating for viable
counts....................................................................... 48
Plating for drug
resistance.................................................................... 49
Sampling for DNA.................................................................................. 49
Sorbitol Solution................................................................................... 49
Sampling for RNA.................................................................................. 49
Cleanup................................................................................................ 51
Counting colonies................................................................................. 51
Media Replacement.................................................................................. 51
Data Analysis.................................................................................... 54
Harvesting.......................................................................................... 55
Setup for harvesting................................................................................ 55
Harvesting............................................................................................... 56
Sample Processing........................................................................... 59
Filtrates................................................................................................... 59
Culture revival......................................................................................... 59
DNA prep................................................................................................. 60
RNA prep.................................................................................................. 60
Lysis buffer........................................................................................... 62
RNA prep for 5 ml daily
samples............................................................ 63
RNA prep for 100 ml
harvest................................................................. 64
Problems............................................................................................. 65
Chemostat References.................................................................... 67
Supplies and Suppliers.................................................................. 68
ATR....................................................................................................... 68
Fisher.................................................................................................... 68
GE Osmonics......................................................................................... 69
Ace Glass.............................................................................................. 70
Masterflex/Cole Parmer........................................................................ 70
R-Biopharm AG...................................................................................... 71
McMaster-Carr...................................................................................... 71
Figures
Figure 1. Testing the limitation in batch.................................................... 8
Figure 2. Testing the limitation in the
chemostat...................................... 9
Figure 3. Tubing and fitting menagerie................................................... 15
Figure 4. Foil origami............................................................................... 17
Figure 5. Assembled media carboy......................................................... 18
Figure 6. Assembled chemostat vessel................................................... 30
Figure 7. Rubber band pump trick........................................................... 35
Figure 8. Status screen........................................................................... 36
Figure 9. Fermenter menu...................................................................... 36
Figure 10. Edit profile 0........................................................................... 37
Figure 11. Edit profile 1........................................................................... 38
Figure 12. Inoculated chemostat............................................................ 41
Figure 13. Header index card.................................................................. 44
Figure 14. Sampling index card............................................................... 44
Figure 15. Small filtering apparatus........................................................ 50
Figure 16. Media conservation trick........................................................ 52
Figure 17. Large filter apparatus............................................................ 56
Planning the experiment
The first thing to do is design your experiment. You need to choose a strain, a limitation, a limiting nutrient concentration, and a dilution rate. You also need to arrange to use the chemostats.
Signup
Once you've got an experiment planned, sign up on the board by the chemostat room. That way, other people can plan their own chemostat use.
Strains
The strains commonly used in the lab are FY, which is an S288C derivative that's been made GAL2+, and CEN.PK, a favorite of the European chemostat community. Using a prototroph is vastly preferred to using an auxotroph. With auxotrophs, you can never really be sure what the cells are using as a source of limiting nutrient. It just complicates matters and makes you less sure of any results. We have prototrophs of FY and CEN.PK, as diploids and as haploids of both mating types, in the strain collection:
|
Background |
Mating type |
AKA |
|
|
FY |
a/alpha |
FY4xFY5 |
MD collection |
|
FY |
a |
FY4 |
11069 |
|
FY |
alpha |
FY5 |
11070 |
|
CEN.PK |
a/alpha |
|
9500 |
|
CEN.PK |
a |
|
11092 |
|
CEN.PK |
alpha |
|
11093 |
The FY haploid strains are from Fred Winston. The CEN.PK diploid, DBY9500, is direct from Peter Kotter. The FY diploid and the CEN.PK haploids were derived in my lab by mating and dissection, respectively.
Both strain backgrounds grow well in glucose, phosphate, and sulfur limitation. I have had very odd results with FY in nitrogen limitation, although CEN.PK seems to behave. All S288C derivatives have a Ty element in HAP1 that decreases its activity. We now also have a HAP1+ derivative of FY from Fred Winston's lab. CEN.PK strains have a mutation in CYR1. Also, LEU2 may not be in the usual location.
You can, of course, use other strains. The biggest unknown danger of a new strain is its flocculation capacity. Because they can stick to the vessel and sink to the bottom to avoid being diluted out, clumpers are selected for in the chemostat. In addition to complicating cell count data, they also make it very difficult to understand what's going on in terms of selection pressure, clonal selection, etc., so the chemostat run is effectively over once they appear. For CEN.PK and FY, I've gotten clumpers occasionally ~400 generations into the evolutions. Many lab strains carry knock outs of several FLO genes, making the transition to flocculation difficult (although some of the knock out mutations, like the one in FLO8 in S288C, are point mutations that may revert). Other strains, such as SK1, flocculate even in rich media, making them next to useless in the chemostat. When using a new strain, be particularly vigilant about frequently checking the culture with the microscope before and after sonication. If sonication effectively breaks up the clumps, I don't think it's a serious enough problem to halt the chemostat, although I would make a note of the phenotype in my log. For short-term cultures, this is not nearly as much of a problem, so you have a wider variety of strains available.
If you do have to use an auxotroph, be very careful with the supplements you add. For example, you can't use adenine sulfate with sulfur limitations. You want to make sure the culture does not become limited for the additive, but you don't want to add so much excess that the culture eats the additive instead of the nominal limiting nutrient. See the Limitations section for how to check limiting nutrients. You can also use an auxotroph on purpose and limit with the additive it requires. Matt Brauer and Alok Saldahna have successfully done this with leu2 strains, and Alok and I have also done ura3 strains. I've included these media formulations in the Media Recipes section.
Limitations
The limiting nutrient depends on what your experiment is. Keep in mind that glucose limited cultures seem to be most sensitive to changes in the dilution rate. Lower dilution rates provoke more respiration while higher dilution rates favor fermentation. Nitrogen limitation does not work well with FY.
If you are not using one of the standard recipe/strain combinations that I list in the Chemostat Media section, you should do a preliminary experiment to figure out the limiting concentration to use. These experiments are conveniently done in batch. Inoculate an overnight culture. Spin it down and resuspend at a 100X dilution in chemostat media without any limiting nutrient. Aliquot equal volumes into a series of appropriate volume shake flasks that contain different quantities of the limiting nutrient. Be careful that the volumes of limiting nutrient solution are the same in all the flasks so you don't get different dilution factors. You may want to make your media 1.1X and bring them to 1X with the limiting nutrient solution. Let these flasks shake at 30C for a couple of days or until the density stabilizes. You want them to be completely in stationary phase. Measure the densities. If you graph the concentration of limiting nutrient vs. the final densities of the cultures, you should get a plot with a linear range, a nonlinear range, and a plateau. You want to stay in the linear range. Here's representative data from one of my experiments for phosphate limitation:
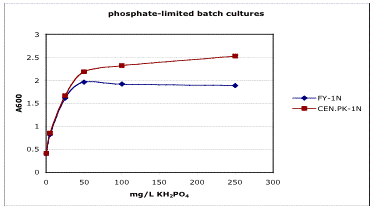
Figure 1. Testing the limitation in batch.
Note that the Y-intercept is not zero. That's probably from nutrient the yeast stored from the overnight culture, which was in rich media in this experiment. Take that into account when you decide on a chemostat media concentration that will yield your desired density. I like to run my evolution starting from a population size of 3-5x107 cells/ml. To measure the final density more exactly, once you have a good idea of what concentration is limiting, grow the first overnight in limiting chemostat media. The cells will use up all the limiting nutrient and the zero will really be zero.
Although the batch results generally match the chemostat quite well, make sure to test the concentration in the chemostat to double check that it's really limiting. To convince yourself that your chemostats are limited by what you think they are, run 2 chemostats to steady state. Once they've hit steady state, in one chemostat, increase the nominal limiting nutrient in the feed media by 50% and watch for an increase in density. In the other, increase the additives by 50% and see if the density changes. If you are truly limited only for the limiting nutrient, you will see ~50% increase in density in the first one but no change in density in the second one. Density is actually not the greatest indicator since it's really yield you're interested in, but it usually does track pretty well. Klett seems to work the best as a surrogate for yield.
In this example from David Hess, six cultures were grown to
steady state with 20 mg/L potassium phosphate. At the indicated point, the feed media was switched to 30
mg/L potassium phosphate. F13,
F15, and F17 are all limited by phosphate. F14 and F16 are not.
F18 washed out.
20 mg/L 30 mg/L
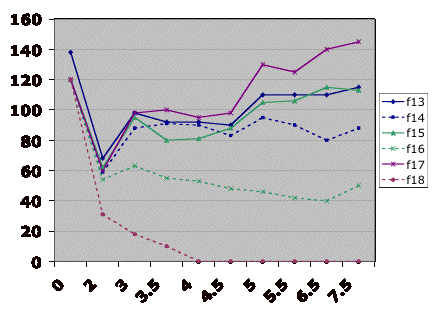
![]() Figure 2.
Testing the limitation in the chemostat.
Figure 2.
Testing the limitation in the chemostat.
Also note that not all strains behave exactly the same. In some circumstances, it may be easier to run two separate chemostats, each with a different (supposedly limiting) concentration of the nutrient. Then compare the steady state densities. This experimental design is particularly useful when the strains take a long time to hit steady state and so evolution is a concern.
Dilution Rate
I have stuck with an arbitrary dilution rate of about 0.17 hr-1. The Paquin and Adams experiments were all at 0.2 hr-1. You will know if you set the dilution rate too high (i.e., above the maximal growth rate) because your culture will wash out. In glucose limitation, there is a critical dilution rate where the culture switches from respirofermentative growth to primarily fermentative growth. Growth-rate dependent changes are currently being studied in detail by the Botstein lab.
Chemostat media
Chemostat media has several components that need to be made separately: salts, metals, vitamins, and carbon source. For each batch of media, you will prepare a carboy, make 1X salts, autoclave the 1X salts, and add the remaining media components via a filter.
These media recipes come from Julian Adams via Frank Rosenzweig with further modification by me. The glucose limitation recipe is exactly per Adams. I modified the glucose limitation recipe for phosphate, sulfur, and nitrogen limitation. In general, I tried to keep all ions at the same molarity where possible. The Adams version of the phosphate limitation recipe uses the salts at 0.25X to limit the effects of phosphate contamination from the other salts, but I always use 1X salts for everything. You can only get away with this if you use really pure chemicals.
The Adams recipe handed down to me also had a typo in it, which we didn't catch until just this year. The potassium concentration was 10X lower than it should've been. If you used my phosphate-limited media recipe prior to December 2005, that recipe was wrong! I'm very sorry about that.
The uracil and leucine limitation recipes use the Adams glucose limitation base plus limiting concentrations worked out by Alok Saldahna.
Salts
Salts can be made as 10X stocks in milliQ water and kept at room temperature until use. Nonsterile salts should be used within a couple weeks of making them to avoid contaminant growth. You may be tempted to make a big carboy of salts, but that experiment has been tried and mysterious floating bits appear eventually. If you want to keep them longer, they can be autoclaved.
Make salts using the purest chemicals available. It is crucial that limiting nutrient concentration not vary due to contamination in other salts.
Note that the phosphate, sulfur, and nitrogen limitation salts do not contain any of the limiting nutrient. I typically put this in with the other additives after the salts are autoclaved.
10X salts for carbon, leucine, or uracil limitation
(1 L)
|
1 g |
calcium chloride.2H2O |
|
1 g |
sodium chloride |
|
5 g |
magnesium sulfate.7H2O |
|
10 g |
potassium phosphate monobasic |
|
50 g |
ammonium sulfate |
10X salts for phosphate limitation
(1 L)
|
1 g |
calcium chloride.2H2O |
|
1 g |
sodium chloride |
|
5 g |
magnesium sulfate.7H2O |
|
50 g |
ammonium sulfate |
|
10 g |
potassium chloride |
10X salts for sulfur limitation
(1 L)
|
1 g |
calcium chloride.2H2O |
|
1 g |
sodium chloride |
|
4.12 g |
magnesium chloride.6H2O |
|
40.5 g |
ammonium chloride |
|
10 g |
potassium phosphate monobasic |
10X salts for nitrogen limitation
(1 L)
|
1 g |
calcium chloride.2H2O |
|
1 g |
sodium chloride |
|
5 g |
magnesium sulfate.7H2O |
|
10 g |
potassium phosphate monobasic |
Metals
Metals are made as a 1000X stock that keeps at room temperature for at least a year. Keep the bottle well wrapped in foil since some of the metals are light sensitive. Make the metals in sterile milliQ. We have not had any contamination issues with this setup. Shake before using since the metals will not totally dissolve.
1000X metals
(1 L)
Dissolve chemicals in ~1 L milliQ in the following order:
|
|
|
Chemical storage |
|
500 mg |
boric acid |
RT shelf |
|
40 mg |
copper sulfate.5H2O |
RT shelf |
|
100 mg |
potassium iodide |
RT, dark, dessicator |
|
200 mg |
ferric chloride.6H2O |
RT shelf |
|
400 mg |
manganese sulfate.H2O |
RT shelf |
|
200 mg |
sodium molybdate.2H2O |
RT shelf |
|
400 mg |
zinc sulfate.7H2O |
RT shelf |
Bring total volume to 1 L with milliQ water.
Vitamins
Vitamins are also made as a 1000X stock. The solution is aliquoted into 50 ml Falcon tubes and stored at -20C. Don't fill the tubes to the top, or else the lid will split when frozen. The "working tube" can be stored at 4C. The vitamins will not dissolve completely, so shake before use. Care should be taken to keep the solution well mixed while aliquoting.
1000X Vitamins
(1 L)
Weigh all chemicals and add to a beaker. Dissolve them as much as possible in 1 L milliQ water.
|
|
|
Chemical storage |
|
2 mg |
biotin |
4C |
|
400 mg |
calcium pantothenate |
4C |
|
2 mg |
folic acid |
RT, dark, dessicator |
|
2000 mg |
inositol (aka myo-inositol) |
RT shelf |
|
400 mg |
niacin (aka nicotinic acid) |
RT shelf |
|
200 mg |
p-aminobenzoic acid |
4C |
|
400 mg |
pyridoxine HCl |
RT, dark, dessicator |
|
200 mg |
riboflavin |
RT shelf |
|
400 mg |
thiamine HCl |
RT, dark, dessicator |
Tubing and Fittings
For reference, here's a guide to the different tubing and connectors and clips I use in setting up the carboys and chemostats:
pump tubing with zip tie T
connector nonlocking locked male Luer
fittings female big clip precut
pump tubing small clip wide bore medium
bore small bore
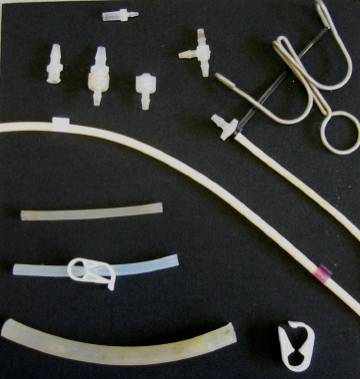
Figure 3. Tubing and fitting menagerie.
The small and medium bore tubing are more or less interchangeable. There's another size between medium and wide that is not pictured. It's useful for setting up the air bubblers.
Carboy
The media vessel is made up of the glass carboy, a big stopper, a filter, a port, and two drop tubes. One drop tube should have a short (~6 inches) length of medium bore tubing attached. This tubing will be used to filter the additives into the autoclaved 1X salts. The other drop tube should have a long (several feet, on the order of the distance from the media vessel to the pump apparatus in the final setup) length of small or medium bore tubing with a male luer lock fitting on the end. This tubing will deliver media to the chemostat. The filter is attached by a ~2 inch piece of wide bore tubing. Make sure the filter does not fall over, cutting off airflow to the inside of the carboy. The filters need to be replaced occasionally.
If you have access to a shop, you can make your own hardware for a carboy. This is considerably cheaper than buying the carboy with the hardware. All you need is #12 stoppers (see supply guide), a sharp cork borer, and some metal tubing cut to size. You will need a thin media supply drop tube that goes all the way to the bottom of the carboy, a shorter length of thin tubing to use for filtering the additives, and a short piece of wider tubing to attach the big venting filter. You may also want to polish the cut ends since the barbs can rip your tubing. This arrangement should be autoclaved with the cork unplugged to allow proper venting. Alternatively, you could put an additional pluggable port in the cork.
Autoclaving the salts
Media is typically made in 10 L batches. 10 L of media supplies a 300 ml chemostat running at a dilution rate of 0.17 hr-1 for about 6 days. Once made, it is stable at room temperature, although Matt has recently observed that the glucose will settle out over a couple of weeks. To solve this, mix the media before using.
Wash out a 13 L glass carboy, hardware, and tubing with DI water. Add 9 L milliQ water. The milliQ machine's metered delivery seems to be adequate for this when frequently calibrated. Add 1 L 10X salts. Do not add amino acids or nucleotides since they may be labile. Add them with the other additives, after autoclaving. Screw down the stopper and mix well. Adjust the metal drop tubes until they sit vertically in the stopper, with the bottoms just at the bottom of the carboy. If the drop tubes are tilted or propped against the bottom, you will stretch out the stopper and it eventually won't hold a vacuum. Use the metal clips to clamp off both tubes near where they meet the metal hardware. This prevents 1X salts from filling the tubing, and thus delivering incomplete media for the volume of the tube. Remove the stopper from the port. Cover the port with foil, taping with autoclave tape if you wish. Cover the ends of all the tubes with foil as well. Wrap the port stopper with foil.
5 4 3 2 1
![]()
![]()
![]()

Figure 4. Foil origami.
One nice way to wrap the tubing ends, taught to me by Frank Rosenzweig, is diagrammed above. It is secure, yet can be undone with one hand.
1. Fold a 4 in x 4 in piece of foil in half top-to-bottom and insert the tubing.
2. Fold it in half again left-to-right.
3. Fold the flap back in on itself so the edges meet the tubing.
4. Fold the top corner on the diagonal to lock the end in the packet.
5. If desired, apply autoclave tape to the foil.
When you go to access the tubing end, you can unfold back to step 2 or 3 with the foil loosely covering the end. It will be protected while you arrange the other piece of tubing.
The locking action doesn't work as well on a tubing end without a fitting (such as the effluent tubing). Be more careful around those since it's easy for the foil to slide off and expose the tubing.
The assembled and foil-covered carboy should look like this:
clip 2 clip 1 Drop tubes Feed tubing Big filter Filtration port

Figure 5. Assembled media carboy.
Several bottles can be autoclaved at once. Make sure to put the autoclave probe in a beaker of water. Autoclave on the liquid cycle for 1 hour. The entire cycle will take on the order of 2-3 hours. Remove the media promptly when the cycle finishes and replace the port stopper. Let cool overnight before mixing in the additives.
Additives
In a small bottle, mix together the carbon source, limiting nutrient, vitamins, and metals, and any other required additives. The following recipes are the standard limitations. You can of course vary the concentrations and the nutrient sources.
glucose (dextrose) limitation additives
(for 10 L media)
For the most consistent glucose limited media, dissolve 8 g glucose (for a final media concentration of 0.08%, 0.24% is also commonly used) in 100 ml milliQ water and autoclave 15 minutes on the liquid cycle. To one bottle, add:
|
10 ml |
1000X vitamins |
|
10 ml |
1000X metals |
phosphate limitation additives
(for 10 L media)
|
10 ml |
10 g/L potassium phosphate monobasic (to 10 mg/L) |
|
125 ml |
40% glucose (to 0.5%) |
|
10 ml |
1000X vitamins |
|
10 ml |
1000X metals |
sulfur limitation additives
(for 10 L media)
|
15 ml |
2 g/L ammonium sulfate (to 3 mg/L) |
|
125 ml |
40% glucose (to 0.5%) |
|
10 ml |
1000X vitamins |
|
10 ml |
1000X metals |
nitrogen limitation additives
(for 10 L media)
|
8 ml |
50 g/L ammonium sulfate (to 40 mg/L) |
|
125 ml |
40% glucose (to 0.5%) |
|
10 ml |
1000X vitamins |
|
10 ml |
1000X metals |
leucine limitation additives
(for 10 L media)
|
15 ml |
10 mg/ml leucine (to 15 mg/L) |
|
125 ml |
40% glucose (to 0.5%) |
|
10 ml |
1000X vitamins |
|
10 ml |
1000X metals |
uracil limitation additives
(for 10 L media)
|
25 ml |
2 mg/ml uracil (to 5 mg/L) |
|
125 ml |
40% glucose (to 0.5%) |
|
10 ml |
1000X vitamins |
|
10 ml |
1000X metals |
Making a carboy of media
Remove a filter apparatus (Nalgene 115 ml 0.45 micron filter unit) from the wrapping. Without contaminating the ends, remove the plastic nozzle where the vacuum would normally attach. Using a sterile pipet tip, pop out the piece of foam blocking the nozzle. Replace nozzle on filter apparatus. Remove foil from the end of the short tube on the carboy. Carefully push the tubing over the nozzle of the filter. Attach the hose from a vacuum to the big filter on the carboy. Start the vacuum and open the clip for the short tubing. Holding the filter at an angle so a sliver of filter remains dry, pour the additive mix through the filter. Once it has all filtered through, tilt the filter so the additives flow into the carboy. If you accidentally wet the entire filter, this step will take a long time, but it will eventually finish. Pour 50 ml milliQ water into the bottle you mixed the additives in. Swirl the water around to dissolve any remaining droplets. Filter as before. Swirl the filtered water around to capture any droplets in the bottom reservoir of the filter apparatus. Again let the mix flow into the carboy. Clip off the tubing. Disconnect the vacuum and discard the filter. Mix the media thoroughly. Matt has noticed problems with the glucose settling out over time. Stirring the carboy on the great big stir plate for an hour right before use seems to help. If you plan to do this, remember to add a big stirbar to the carboy before autoclaving.
Alternative Filtering Strategy
We've recently switched to filter-sterilization. It seems to make more consistent media, and you cut down on autoclave accidents. Autoclave an empty carboy. Mix up all the media in a separate carboy and stir well to dissolve everything (you can use salts, vitamins, and metals from stock solutions, but you should use granulated glucose). Attach a bottle top filter with the cotton filter removed (see suppliers at end) to a 100 ml bottle. Attach to the filtration port in the carboy, hook the vacuum up to the big top filter, and filter it all in. This will take ~15 minutes. Although the filters are nominally for only 1 L, we have not had any contamination problems associated with filter failure.
Attaching the carboy
Right before attaching the media, prime the tubing leading to the chemostats. Remove the clip from the top and blow into the carboy through the big filter. Once you see media traveling though the tube, prepare to clamp the end. The goal is to fill the tubing with media without spilling any. If media does come out the end, douse the completed assembly junction with alcohol or 10% bleach to limit contamination.
Lift the carboy onto the shelf above the chemostats. There are a few lifting strategies favored, so ask around. If you feel at all unsafe lifting the media yourself, ask someone to help. If you are connecting multiple vessels together, make sure they are on the same height shelf.
Remove the foil from the end of the tubes you are connecting and, without contaminating them, twist the luer locks tightly together. Remove the clip to start media flowing.
See Changing the Media later in the manual for further instructions.
Plates
You will need YPD and minimal plates for the viable cell counts, and drug selection plates for the sweep monitoring.
YPD
(0.5 L)
Dissolve the following in ~400 ml milliQ water:
|
5 g |
Bacto yeast extract |
|
10 g |
Bacto peptone |
pH to 6.
Bring the volume up to 500 ml. Pour into a 1 L bottle.
To make plates, add:
|
10 g |
Bacto agar |
Swirl and autoclave for 15 minutes on the liquid cycle. Make sure to put the autoclave probe in a beaker of water. Also, put the bottles in a pan to contain any spills. Carefully remove from the autoclave, let cool slightly, and swirl to distribute the agar. Use immediately or let solidify. Tighten down the caps.
Melt the agar in the microwave, a few minutes at a time, with frequent swirling. Put in a 55C waterbath to cool.
When you can handle the bottle, add
|
25 ml |
40% glucose |
|
5 ml |
5 mg/ml adenine sulfate |
|
4 ml |
5 mg/ml tryptophan |
Mix well.
YNB
(0.5 L)
Dissolve in 0.5 L milliQ:
|
3.35 g |
Yeast Nitrogen Base without amino acids |
Pour into a 1 L bottle.
To make plates, add:
|
10 g |
Bacto agar |
Swirl and autoclave for 15 minutes on the liquid cycle. Make sure to put the autoclave probe in a beaker of water. Also, put the bottles in a pan to contain any spills. Carefully remove from the autoclave, let cool slightly, and swirl to distribute the agar. Use immediately or let solidify. Tighten down the caps.
Melt the agar in the microwave, a few minutes at a time, with frequent swirling. Put in a 55C waterbath to cool.
When you can handle the bottle, add
|
25 ml |
40% glucose |
Mix well.
Supplements for YNB
Cold Spring Harbor manual plus modifications by Katja Schwartz
|
|
stock, mg/ml |
ml/0.5 L media |
Stock Storage |
|
adenine sulfate |
5 |
5 |
RT |
|
uracil |
2 |
5 |
RT |
|
tryptophan |
5 |
5 |
4C |
|
histidine-HCl |
5 |
2 |
4C |
|
arginine-HCl |
10 |
1 |
4C |
|
methionine |
5 |
2 |
4C |
|
tyrosine |
2 |
7.5 |
4C |
|
leucine |
10 |
5 |
4C |
|
isoleucine |
10 |
1.5 |
4C |
|
lysine-HCl |
10 |
5 |
4C |
|
phenylalanine |
10 |
2.5 |
RT |
|
glutamic acid |
10 |
5 |
RT |
|
aspartic acid |
10 |
5 |
RT |
|
valine |
37.5 |
2 |
4C |
|
threonine |
40 |
2.5 |
RT |
|
serine |
80 |
2.5 |
4C |
canavanine plates
(0.5 L)
Make the YNB agar as above. When it cools slightly, add
|
0.7 g |
arginine dropout powder |
Swirl to dissolve.
Let cool until comfortable to handle. Add
|
25 ml |
40% glucose |
|
1.5 ml |
20 mg/ml canavanine sulfate (in milliQ, filter sterilize stock and store at 4C) |
Mix well.
Dropout powder
|
1 g |
adenine sulfate |
|
1 g |
uracil |
|
4 g |
L-tryptophan |
|
1 g |
L-histidine |
|
1 g |
L-methionine |
|
3 g |
L-tyrosine |
|
4 g |
L-leucine |
|
4 g |
L-isoleucine |
|
3 g |
L-lysine |
|
2.5 g |
L-phenylalanine |
|
5 g |
L-glutamic acid |
|
5 g |
L-aspartic acid |
|
7.5 g |
L-valine |
|
10 g |
L-threonine |
|
20 g |
L-serine |
|
2 g |
alanine |
|
2 g |
cysteine |
|
2 g |
proline |
|
1 g |
L-arginine |
Grind up in a blender until very fine and even. Use 0.7 g/0.5 L media.
If you wish to make an arginine dropout mix, leave out the arginine. To make a ura-leu-his-lys-ade-trp drop out mix, leave out those 6 chemicals and use 0.6 g/0.5 L media.
5-fluorouracil plates
(0.5 L)
Make YNB agar as above. When cool enough to handle, add
|
25 ml |
40% glucose |
|
0.5 ml |
26 mg/ml 5-fluorouracil (in DMSO, store RT wrapped in foil) |
Mix well.
alpha-aminoadipate plates
Cold Spring Harbor yeast course manual
(0.5 L)
|
0.8 g |
yeast nitrogen base w/o amino acids or ammonium sulfate |
|
10 g |
Glucose |
|
15 mg |
Lysine (30 mg/L) |
|
10 g |
Bacto-agar |
|
480 |
Milli-Q water |
Dissolve the first 4 ingredients. Add the agar. Autoclave for 15 minutes on the liquid cycle, with the probe in water. Once it has cooled enough to handle, add 20 ml 5% alpha-aminoadipate solution:
|
1 g |
Alpha-aminoadipate (aka adipic acid) |
|
20 ml |
Milli-Q water |
Mix and adjust pH to 6 with 10 N potassium hydroxide to allow dissolution. Filter sterilize and add to autoclaved ingredients.
Kanamycin (aka G418 or Geneticin) plates
From Sandy Silverman
(0.5 L)
Make YNB agar as above. When cool enough to handle, add
|
15.15 ml |
20 mg/ml Geneticin |
|
25 ml |
40% glucose |
The Geneticin can be dissolved in water, filter sterilized,
and stored at 4C. Keep in mind
that Geneticin is not completely pure.
This recipe is based on a bottle that was 755 µg Geneticin/mg. Sandy has observed occasional
variability using this recipe.
Increasing the Geneticin by 50% seems to help.
The Chemostat Vessel
For each chemostat, you have a culture vessel, O-ring, stirrer/probe/aeration/port assembly, clamp, effluent drop tube, various probes, pump tubing, air inflow, air release, inoculation port, and effluent tubing.
Assembling the vessel
Even if it looks like the last person to use the vessel cleaned it, rinse it again with DI water before you use it. Never use soap on any chemostat item. Keep the little plug over the temperature probe when washing and autoclaving.
Make sure the stirrer is snug in the housing. If it is not, snap it into place. Push the little o-ring against the joint to hold the stirrer in place. Place a big o-ring in the rim of the glass vessel. Put the stirrer assembly into the vessel. Open up the clamp, fit it around the neck of the chemostat, and clamp down until it no longer moves easily. Make sure the clamp is seated properly on the rim of the vessel. Put a bumper (fat black rubber o-rings) around the neck of the vessel. Set the assembled chemostat in the carrying rack.
Determine which probes you want to include. We currently have foam, pH, and dissolved oxygen. Unscrew the appropriate size plugs and screw the probes into the ports on the top of the vessel. Remove the plastic caps from the probes. Set the plugs and caps aside in the Hardware drawer so we can find them later. The pH and DO probes must always be kept in water, buffer, or media. When not in use, they should be capped and placed in the rack with the others.
Use the marked clear plastic ruler to calibrate the working volume. We typically run the chemostats at 300 ml. The markings on the ruler are for the height of the top edge of the effluent tube relative to the top of the casing it sits in. The height depends on the number of rotors and the number and type of probes. To adjust the height of the tube, unscrew the small screw on the side of the casing. Adjust the tube to the appropriate height and tighten the screw back down.
If you are using the oxygen probe, don't adjust the effluent tube height to the working volume. Instead, push the tube almost all the way to the bottom of the vessel. This maintains sterility when you go to eject the water before you start the chemostat.
If you are using a novel combination of vessel, rotors, probes, and desired working volume, you will need to calibrate the volume yourself. The easiest way to do this is to set the vessel up how you want it, and fill it with the desired working volume plus 100 ml extra (measure carefully) water. Pull the effluent drop tube out of the water. Start the chemostat running at the same aeration and mixing rate you plan to use. Put the end of the effluent tube in a 100 ml graduated cylinder. Now slowly lower the drop tube until some of the water ejects. Continue to do this until you have about 95 ml in the graduated cylinder. Leave it alone for a few more minutes and see if any more ejects. Generally, a few more mls will come out. The goal is to get exactly 100 ml to eject. Sometimes you'll have to try it a few times to get it right on, but it's worth the extra effort to get an accurate working volume. Once you are happy with the result, label the ruler at the height of the top edge of the drop tube.
The interior diameter of the pump tubing you use will in part determine the dilution rate of the chemostat. We have two kinds of pump tubing: smaller tubing that gets cut into lengths and precut larger tubing. Refer to the tubing and fitting reference in the Media section for a picture. The precut tubing has stops glued to it to prevent it from getting pulled into the pump. The smaller tubing requires a zip tie to secure it: cut a piece of pump tubing long enough to go through the pump head and to the chemostat. Put a female luer lock fitting on one end. Use a zip tie to secure the pump tubing on the barbed end of the fitting. Attach the other end of the pump tubing to one of the ports on the chemostat. If using the precut tubing, attach one end to a port and a female luer lock fitting to the other. The stops on the precut tubing occasionally come off. If this happens to you, secure the tubing around the barbed end of the fitting with a zip tie. It seems to go by batches, so if a batch seems to have weak stops, add a zip tie just in case.
Attach a 1.5 ft piece of medium bore tubing to the effluent tube. This should be long enough to reach the effluent vessel in front of the chemostats.
Attach a ~3 in piece of medium bore tubing to the air input port. Put a non-locking luer fitting on the end.
Attach a ~3 in piece of small or medium bore tubing to another port. This will be the inoculation port. Slip a small plastic tubing clip over the tubing. If you like, you can add a female luer lock fitting on the end.
For an optional secondary air release port, attach a ~3 in piece of small or medium bore tubing to another port. Slide a small plastic tubing clip over it. Put a non-locking luer fitting on the end.
Any free ports need to be plugged. You can make plugs out of scrap ~1 in small or medium bore tubing with silicone glue injected in one end. A less desirable option is short pieces of tubing with clips.
Keep tubing clips open during autoclaving to allow steam venting. If the vessel is full of water, clip the effluent tube and the air tube to prevent ejecting the water.
Cover all the tubing ends with aluminum foil. Put a piece of autoclave tape over some of the foil if desired.
It should look more or less like this:
Inoculation port clamp Effluent tube Air input Probe Pump tubing

Figure 6. Assembled chemostat vessel.
Also, decide how you will arrange the media flow to the chemostats. If you want to run multiple chemostats off one media carboy, you need to make a Y connector out of scrap tubing. Make sure you put the right connectors on all the ends. If you want to run chemostats off multiple media carboys, you will need a more complicated branching connector. Make the connector piece and wrap foil over all the ends. Don't hook it to the chemostats yet since it will interfere with setting them all up.
Autoclaving the chemostats
If you are running chemostats with oxygen probes, fill the chemostats with water or media and autoclave them for 15 min on the liquid cycle. Make sure to add enough liquid that the probes do not dry out. Also, put the autoclave probe in a beaker of water.
If you are autoclaving the chemostats dry, use the bedding setting for 15 min.
Close all the tubing clips once the chemostats come out of the autoclave.
Small air filters should be individually wrapped in foil and autoclaved for 15 minutes on the bedding cycle.
Extra effluent tubing lengths should have both ends wrapped in foil before being autoclaved for 15 minutes on the bedding cycle.
Setting up a run
Assemble the sterile chemostats and media vessels for your run. You may want to start setting up the day before you inoculate, especially if you are using the dissolved oxygen probe. You need to start the air supply, connect the media, connect the probes, fill the vessel with media, and program it. Then you can inoculate.
It takes 5 days to run a standard short term chemostat for harvest. A convenient schedule might be:
|
Sunday: |
polarize oxygen probe, inoculate overnight culture |
|
Monday: |
setup and inoculate in batch mode |
|
Tuesday: |
pumps start |
|
Wednesday: |
measure effluent and check dilution rate |
|
Thursday: |
at or near steady state, full sampling |
|
Friday: |
at steady state, full sampling, ready to harvest |
Obviously, if your dilution rate is much different from 0.17 hr-1, your timing will vary. The 5 day scheme is ~15 generations. Much less and you're not reliably at steady state. Much more and you have evolution to worry about. Some strains take longer to reach steady state. Do sufficient sampling to convince yourself that the chemostat really is at steady state before you harvest.
Polarizing the dissolved oxygen probes
The oxygen probes must polarize for at least 6 hours before you begin. Put each chemostat into a holder on the machine. Plug the dissolved oxygen probe into the gray cord by pushing down and twisting the connector. It should lock into place. Make sure the main power to the machine is on.
Setting up the vessel
Unscrew the casing on the pump head. Lift up the black plastic part and thread the pump tubing through the little metal catch in the back. Pull the tubing straight out from the catch and through the opening in the black plastic part. Screw the pump head all the way back down.
Connect any Y connectors by twisting the luer lock fittings tightly together.
Connect the primed media vessel tubing (see Media Preparation section) to the pump tubing by twisting the two luer lock fittings tightly together. Remove the clamp from the media feed tubing. If you've properly secured the pump head, no media should flow into the vessel.
Get an autoclaved 2 L Erlenmeyer flask. Punch a hole in the foil cover with a pencil. Remove the foil from the effluent tubing and insert the end into the flask.
Plug the red temperature cord into the plug. The red dots should line up. On the newer model, the metal probe on the red cord should be threaded into the metal casing on the vessel (Important! Unlike on the old machine, which assumed a high temperature if you forgot to plug in the probe, with the new probe, if you forget to attach it, the machine will read room temperature even though the heating element is on, cooking your culture).
If you're running <30C, or at several different temperatures on one sixfors, use one of the machines with a chiller. The chiller needs to be refilled every few days.
Connecting the air
Make sure the air bubblers are filled with water. If necessary, top them off with milliQ water. The top of each bubbler should be held on by 4 rubber bands, 2 on each side, or 2 springs. If any of these have snapped, replace them.
Unwrap an autoclaved air filter and the end of the air input tubing. Snap the large end of the filter onto the luer fitting. Trace the tubing from the air pressure gauge past the air bubbler to find the end. Slip the tubing over the small end of the filter.
Open up the airflow at the gauge. Don't worry; you cannot blow anything up by opening it too fast. When you see the little ball start to float, dial the airflow down so the bottom of the ball is just hovering above the bottom of the gauge at the 6 marking. Check the bubbler and the vessel to make sure air is flowing. If the vessel was filled with water, the overflow should start ejecting from the effluent tube. If it seems like air is escaping somewhere, make sure the top of the bubbler is secure. Other possible problems are unclipped ports, broken clips, and misaligned O-rings. The last problem can sometimes be fixed by tightening the clamp around the top of the vessel.
Starting the media flow
If you are using the dissolved oxygen probes, you'll need to eject all the water/media in the vessel. Loosen the little screw on the side of the effluent tube. Lower it to the bottom of the vessel and allow the water to empty. Use the ruler to reset the height of the tube and tighten the screw.
In order to maintain sterility of the media, you must make sure air from the chemostat vessel never flows into the media carboy. The easiest way to do this is to pop the air tubing off the filter whenever you plan to loosen the pump heads. Right now, this possibility isn't such a problem since there are no cells in the vessel, but it's a good habit to get in to and the air pressure interferes with the media flow anyway.
Unscrew the pump head until you see media start to flow into the vessel. While the vessel is filling, you can fiddle with the pump head. The objective is to position the pump tubing just right so that no media flows when the pump is tightened all the way down. You also want the media to flow evenly and immediately when the pump head contacts the pump driver (that shiny rotating bar you see behind the pump heads). It might require repositioning the tubing several times to get this right. Even then, the pump may need more adjusting later. If the vessel fills up past the effluent drop tube while you're adjusting the pumps, clamp off the media flow. If you are having a hard time stopping the flow, try pushing the pump head against the driver. Sometimes that stops the siphon.
If the pump head is squeaking or skipping, and repositioning doesn't help, try wiping the rubber grip with a damp Kim wipe. Back dust tends to accumulate as the rubber degrades and that interferes with the grip. Try switching to a different pump head if all else fails. Another trick is to loop a rubber band from the screw to the metal bars along the back of the machine. This seems to give the pumps a little extra boost.

Figure 7. Rubber band pump trick.
Once you're happy with the pump tubing, let the vessel fill up until the media level is just above the effluent tube, and tighten down the pump head. Reattach the air. The excess media should eject, giving you the correct working volume. If it doesn't eject, check that all your clips are closed, your tubing is intact, and the O-ring is seated properly. Air may be escaping instead of pressurizing the vessel.
Programming the chemostat
Each vessel of the chemostat needs to be individually programmed. The chemostat interface can be counterintuitive sometimes, so be careful and double check all your settings.
Enter usually accepts a new value and Esc usually cancels it. When changing entries, you must hit enter after a numeric entry to accept the change. The +/- key changes a toggle field. For toggle entries, don't hit enter after changing them unless it's the last change you make on the screen. Esc also cancels a menu screen, leading you to the previous menu.
Profiles
For each fermenter, you need a profile or chain of linked profiles to control the fermenter parameters. Typically, you will run the chemostat in batch overnight before turning on the pumps. This involves two linked programs. First, use the esc key to get to the main menu. The esc key will now toggle between the main menu and the status screen, which shows data for all the vessels.
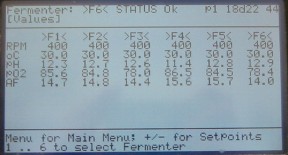
Figure 8. Status screen.
From the main menu, select 1 fermenter menu. You usually shouldn't need to select any of the other options on the main menu. Enter the desired fermenter number at the prompt. You're now in the Fermenter menu.
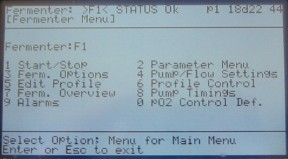
Figure 9. Fermenter menu.
Select 5 Edit Profile. Generally, you will be editing pre-existing profiles. Input the number of the profile you want to edit and hit enter. Profile 0 is usually set up to run 24 hours in batch (i.e. no pumps) with temperature control and mixing. The standard temperature for a yeast chemostat is 30 and the mixing is 400 RPM. Make sure these settings are correct. If you need to edit a setting, use the arrows to scroll to the field and type in the new number. Hit enter to accept the change. For toggle fields, such as turning pumps on and off, you do not need to hit enter to accept the change. Once you have finished the edits, hit enter again to accept all the changes. You should be back in the fermenter menu. Hit 5 to go back to Edit Profiles and make sure your changes were accepted.
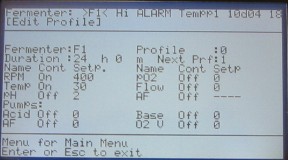
Figure 10. Edit profile 0.
Note that you can only edit one profile each time you go into the Edit Profiles screen. If you try to edit more than that, only your most recently edited profile will actually accept the changes.
You can link programs into chains and loops using the next profile field. After spending 24 hours in profile 0, the chemostat should switch to a new profile that turns on the pumps. Often, that profile will be number 1, but you may use other ones. Just make sure profile 0 correctly links to the appropriate next profile. The next profile can repeat indefinitely by running for any amount of time and then linking to itself as the next profile. The only difference between profile 0 and the next profile is the addition of pump settings. Choose which pump you want to use and scroll to the on/off/auto field. Hit the arrow keys until the pump is on. Scroll to the numerical field for the pump and enter the number of counts the pumps should operate each cycle (refer to table below). You'll set the cycle counts in a different menu. Hit enter to accept the change and enter again to return to the fermenter menu. You may want to return to the edit profiles menu to double check the settings.
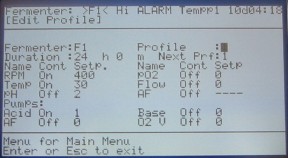
Figure 11. Edit profile 1.
To set the pump cycle, hit 4 pump flow settings. The only part of this window that you need to edit is the cycle. Acid and base pumps must have the same cycle count, so editing one of them updates the other automatically. The AF pump can be on a different cycle. Hit enter to accept any changes and return to the fermenter menu.
The different machines require different settings. Cheryl Christianson calibrated them using the Pharmed collared pump tubing:
|
Machine |
Set counts |
Cycle |
Dilution rate |
|
Old |
1 |
8 |
0.171 |
|
Old |
1 |
7 |
0.183 |
|
Old |
3 |
20 |
0.200 |
|
Old |
2 |
13 |
0.209 |
|
New |
1 |
9 |
0.167 |
|
New |
2 |
17 |
0.195 |
|
New |
3 |
25 |
0.200 |
|
New |
1 |
8 |
0.210 |
|
New |
1 |
7 |
0.242 |
Now, you need to tell the chemostat which profiles to run. To do this, go into 6 profile control. Set actual profile to be the starting profile (i.e., 0) and last profile the final profile in the chain or loop (i.e., 1). Hitting reset profile restarts the timer for the actual profile. This feature will be used below to restart the 24 hour batch countdown once all the vessels are inoculated. If you stop and restart the chemostats, you should check the profile control window to make sure it remembered which profile to use. Sometimes it will change to the mysterious profile -1 instead of reverting to the profile it was running.
Finally, to start the chemostat, go into 1 start/stop from the fermenter menu. Scroll down to start fermenter and toggle it to on. Hit enter. The impeller should start to spin up, and the temperature will start to adjust. To monitor the progress, use the esc key to return all the way up to the status page.
It is usually a good idea to stop the chemostat whenever you need to edit anything. Return to 1 fermenter menu and 1 start/stop. Toggle the field to on and hit enter. Check the profile control before restarting.
Calibrating the Dissolved Oxygen Probe
Once the chemostat is set up and running, but not yet inoculated with cells, you can calibrate the dissolved oxygen probes. DO is measured relative to the minimum reading on the probe obtained under nitrogen sparging and the maximum reading obtained under air sparging. The minimum reading should not change from run to run. To get to the calibration page, first hit esc until you are at the main menu. Hit 1 fermenter menu and select an individual fermenter. Then go: 2 parameter menu -> 4 pO2 -> 5 calibrate all. The screen shows the high and low reference points and the actual reads for each fermenter. You can use whatever scale you like. However, it's easiest to use 0-100%, where 100% is the O2 concentration of room air.
The low reading is typically around 140 on the old machines, and less on the newer machines. If it is much different than expected, recalibrate the low end. At the gauge, disconnect the tubing delivering air to the chemostats. Turn on the nitrogen tank at the regulator until you can feel the nitrogen flowing, but not too strong. Hook the tubing from the nitrogen tank to the bubbler and make sure it makes approximately the amount of bubbles you were getting with the air. On the calibration screen, look at the read for the appropriate fermenter. When it stops changing, scroll to the low read number and hit the number key for the fermenter you're calibrating. Hit enter again to exit the screen and accept the new setting. Go back into the calibrate all menu and make sure the read is the same as the loR for that fermenter. Repeat the process for each fermenter.
Once all the low refs are set, disconnect the nitrogen and reconnect the air. Repeat the entire process, except now using the high reads. It may take quite a while for the solution to fully saturate and the probe readings to equilibrate. Be patient.
Matt has set up the computer to record the DO probe readings. To start data logging, type
./start.pl username x y z
where username is your username (i.e. maitreya) and x, y, and z represent the numbers of the vessels you want to monitor. You can start as many vessels as you like, not just 3. Numbering starts with 1 right next to the sink and goes clockwise to 24. Make sure to record the run number.
Calibrating the temperature probe
We've noticed that the temperature probes tend to drift as much as 1.5 degrees from the supposed temperature. You should recalibrate them once in a while, and every time you're running a chemostat where precise temperature control is important. The calibration procedure is courtesy of David Hess.
Fill a chemostat with 300 ml water. Open one of the ports on the top to insert a thermometer that you trust. Using a good digital thermometer accurate to tenths of a degree C is preferred.
Set a program to run at the conditions you would normally run a chemostat at in batch phase (i.e 400 RPM, pumps off) but set the temperature to 37C. When the screen says the temperature has reached 37C measure the actual temperature with the digital thermometer and record the value. With the chemostat still running, select the fermentor menu and then the chemostat you are calibrating. Once in the specific fermentor menu select 2 Parameter Menu -> 2 Temp -> 2 Calibrate. Scroll down to the Hi Ref value and enter the temperature value measured with the thermometer. This will set the high value.
Repeat Steps 1-4 running the chemostat at 28C and enter the measured value in the Lo Ref entry. The chemostat will now be well calibrated in that temperature range.
Inoculating the chemostats
I prefer to inoculate the chemostat with fresh overnight culture made from a fresh colony. Plan on 2-3 days to grow colonies streaked from a frozen glycerol stock, plus another day for the overnight culture. You can grow the overnight in YPD or the chemostat media. If you use YPD, spin down 1 ml of culture and resuspend it in water.
Find the inoculation port on the chemostat and loosen the foil covering it. Pop the air tubing off the filter to prevent your sample from getting blown out as you add it. Pipet up the cell culture, unclip the tubing, remove the foil, and pipet the culture into the inoculation port by snugly fitting the pipet tip into the tubing end or the fitting. If I am running an evolution, I wash the inoculation port with some water to prevent growth in the port that could recolonize the chemostat. I have no evidence that this step actually helps. Clip the tubing closed and reconnect the air.
Once all the chemostats are inoculated, go into each fermenter menu and enter profile control. Hit reset prf and enter to restart the 24 hour clock. Write down the time. Resetting the profile is one of the few things you can reliably do while the chemostat is running.
Empty all of the effluent vessels of any extra media or water.
The whole thing should look like this:

Figure 12. Inoculated chemostat.
Signup
Update the chemostat signup board once you've started your chemostats. That way if something is amiss about a vessel, everyone knows whom to notify. It's a good idea to leave your phone number, too, especially if someone else is helping you on the weekends and holidays.
Another way to
monitor the chemostats is via the cam-o-stat, a web cam my husband, Mark Huang,
set up so I would no longer need to go to lab in the middle of the night to
check my media supply. The
cam-o-stat is still a work in progress.
We only have one right now, but we'll eventually get the whole room
outfitted.
Sampling
the chemostats
The chemostats ideally should be sampled every day, particularly when collecting sweep data. Try to be as consistent as possible about your technique, and write down anything that you change.
My daily sampling regimen includes making a glycerol stock, looking at the culture under the microscope, sampling for RNA and DNA, and measuring effluent volume, optical density, Klett reading, Coulter count, Coulter mean cell volume, viable cell counts on YPD and minimal plates, and sweep marker frequency on selective plates. Depending on the number of chemostats and plates, the whole process, including setup and counting the plates from earlier in the run, takes anywhere from an hour to as much as 3-4 hours.
When you need to change the media, try to do it after sampling to avoid any chance of a perturbation.
Data collection
It is extremely important that your data be collected in an organized and meaningful way. Charlotte Paquin was able to look up chemostat data for me 20 years after her thesis work. This is the level of organization and clarity you should aspire to.
I use an index card system coupled with Excel spreadsheets. My system has the advantages of being very portable, completely customizable, and available in both hard copy and electronic copy in case disaster strikes. Matt uses a roughly similar system with preprinted record sheets instead of index cards. Choose whatever works for you.
For my index cards, I start with a header card with all the relevant information about the chemostat, including fermenter number, strain, media composition, limitation, date and time started, and method of inoculation. I also make a similar entry in my master chemostat index spreadsheet on the computer.

Figure 13. Header index card.
Each day, I label all the fields on a new card before I start sampling. This includes what dilutions should be done for all the measurements, what plates need to be used, and what was written on the frozen sample. Each chemostat gets a card. As I take all my measurements I fill in the fields. I write down observations and illustrations of the look and smell of the culture on the back of the card. The card then goes into my working box. I pull the cards whenever I can fill in another field, such as colony count or dry weight.

Figure 14. Sampling index card.
Once all the fields are filled in, I file the cards in a small box for active chemostats. Once the chemostat run is completely finished, I fill in the end date on the header card, amend the entry in the chemostat index file with the end date and any pertinent results (i.e., whether or not I harvested and where I put the tubes) and type all the data into the computer. You could also input the data periodically while the chemostats are running. Finally, I clip all the cards together with a secure paper clip and file them in my archives. No cards make it into the archives without getting entered into the computer. Make sure your computer gets backed up regularly. If it isn't, at least burn your data to CD once in a while and store it off-site.
Sample tracking
The combination of fermenter and date should give a unique identifier for sample tracking. Glycerol stocks added to the main strain collection will also have a unique collection number. If you institute any sort of shorthand or alternate naming scheme, it's a good idea to keep track of it in the main chemostat index worksheet. Once you've run a few chemostats, you can get awfully confused about what's what. You may want to keep track of where in the freezer you put all the daily samples and the harvests. I record this information in my index.
Preparing to sample
One nice feature of the card system is that it helps you organize all the required plates and diluents for sampling. You should get all of this set up before you start sampling. Label all the required tubes for the density measurements and serial dilutions and fill them with the right amount of diluent. Label all the plates. Turn on all the equipment and check that they're properly calibrated. Run a clean sample of diluent through the Coulter Counter to make sure it's clear of excess cells.
Prepare the Klett colorimeter. The Klett is nice because it scales very close to linear with dry weight, unlike the other density measures we use. Abuse of the Klett can blow the bulb, which we have to buy from some antique dealer on the web. Read the manual or ask someone if in doubt. Start with the machine turned off. Make sure that a filter is in place, i.e. the switch on the side is on. Put a water or media blank into place. Turn the big knob up front so the scale is at 0. Adjust the pointer to 0 with the small knob on top and center of the Klett (aka "dark" zero). Switch on the light. Readjust zero with the small knob further back on the machine (aka "blank" zero). Recheck the zero right before you use it.
Contamination issues
Contamination of the chemostat culture ends the experiment and casts doubt on the previously collected data. Be aware of the sterility of all chemostat accessories at all times. Media vessels are particularly contamination-prone. Contamination of the media vessel often manifests as a film of cells on the bottom of the carboy. If the contaminant eats all the limiting nutrient, the first symptom may be that your culture washes out. Contamination of the effluent tubing is unavoidable. As far as I can tell, I have not had any contaminants work their way all the way up the effluent tubing and into the vessel. The positive pressure from the air flow seems to be enough to contain it.
You will probably only notice the tubing contaminants if they're drug resistant. Since the vast majority of the cells you collect from the effluent tube are yeast, you should get very few, if any, on a YPD plate. Testing for rare markers can mean plating much larger aliquots of culture, revealing the presence of low-level contamination. If you are monitoring endogenous drug resistance markers for sweeps, drug resistant interlopers will interfere with accurate colony counting. The most foolproof way to get around this is to replace the effluent tubing every day before sampling. Cut a bunch of lengths of tubing, wrap the ends in foil, and autoclave. Replace the old tubing with new sterile tubing right before you sample. If you still have contamination, check for contamination of the main culture by unscrewing one of the ports in the top and taking a sample. Check the sample under the microscope and on plates.
Cells can also migrate into the lower part of the pump tubing from the vessel. We're not really sure how this happens at the moment. Keeping the inoculation port far away from the pump tubing port may help.
Sampling
Once you've replaced the effluent tubing, if desired, it's time to start sampling. Since you will take a Klett reading anyway, the easiest way to collect a sample is to put the end of the effluent tube in a sterile Klett tube, resting the end near the top so air bubbling doesn't make the tube overflow. Write down the time. The Klett requires about 5-10 ml of culture for a good reading. That volume will take a few minutes to collect. While the tube is filling, pour the effluent into a graduated cylinder and write down the volume. You'll use this measurement to calculate the dilution rate later.
Klett
Once you've collected about 10 ml in the klett tube, cap it and mix it by inversion or vortexing. Let any bubbles clear out and wipe any spilled culture off the tube with a kimwipe. Check that the media only blank is still reading 0. Put the tube in the Klett and turn the big knob on the front until the little arrow lines up perfectly with the line on the meter. Record the reading.
Glycerol stock
If you are freezing aliquots of the culture, pipet 1 ml culture into 0.5 ml 50% sterile glycerol in a clearly labeled cryovial. Invert a few times to mix well and put the sample at -80C.
Spectrophotometer
Dilute the sample appropriately in water or media for the spectrophotometer reading. I like to use the same dilution for the entire chemostat run, so I start a little on the low end of the linear range, which is about 0.1-0.5. I generally dilute 0.5 ml culture into 1.5 ml water in a glass tube. Vortex the tube and pour the contents into a cuvette. Fill up the carousel with samples, making sure the water blank is in the right position and that the cuvettes are oriented properly with respect to the light path. Use the cell growth program to read the optical density at 600 nm, editing the program for the number of samples you have. Record the measurements.
Sonicator
Pour ~5 ml culture into a plastic round-bottom 15 ml tube. I am conservative in my use of the sonicator and prefer not to use glass tubes with it. Wear safety glasses and ear covers when you use the sonicator. Check the tip occasionally for cracks and other signs of wear. The tip needs to be replaced every once in a while. The power dial should be set at 12 and the switch on the front at remote.
Wipe the tip of the sonicator with a kimwipe saturated in ethanol. Completely immerse the narrowest part of the tip in the tube of culture. Use the button on the top of the sonicator probe to make 10 ~half second pulses. They should be evenly spaced and of equal length. The sound should not be a high-pitched squeal. You'll get to know the usual behavior of the sonicator after using it a few times. If it deviates from the usual behavior, let someone know. Wipe the tip with the kimwipe. Keep repeating until you've finished all your samples. Clean the tip thoroughly with ethanol and turn the machine off when you are finished.
Coulter counter
The Coulter counter interface is not the most user friendly in the world, and unguided button-pushing can completely screw up the settings. Check the manual or talk to someone if you need help. The machine will usually be in the Instrument functions screen when you start using it. Hit the Output button to change to the Analysis screen. Make sure the dilution setting is correct. Is usually use a 1/1000 dilution. Check that a sample of blank diluent gives a low count on the Coulter counter. Fill a clean cup with ~20 ml fresh IsotonII. Lower the platform and place the cup in the space. Return the platform to the top, and hit start. If the count is higher than about 106 cells/ml, clean the machine by flushing with fresh diluent and then recounting until the count is reasonable.
Once you are satisfied, vortex the sonicated sample. Add 10 µl culture to 10 ml IsotonII in a glass tube. Avoid carrying extra beads of culture on the outside of the pipet tip. Vortex the sample well. Pour it into a cup and place it in the holder. Hit start. Listen for the clicks as it counts. The clicks should be evenly spaced. Look at the window with the view of the sampling aperture. Is should be free of debris. The screen meter that monitors concurrent particles (CONC) should stay constant and low. If any of these conditions are not met, your count will be inaccurate, in which case you should recount the sample. If the sample itself seems contaminated by lint or whatever, make a new 1/1000 dilution in fresh diluent. If debris seems stuck in the aperture, use the Unblock button to try to flush it. You want the total number of particles counted to be about 10000-20000 to get an accurate estimate. You may need to make a different dilution if your sample is more or less dense than normal.
Once you are satisfied with the count, write down the particles/ml. Hit output to go to the next screen. Scroll the stat field until the mean volume is displayed. Write that down. If desired, draw the particle size distribution. You can also print out a sheet with all this information by hitting the print button. When you're finished, hit the Functions button to return to the Instrument functions screen. Place a sample of fresh IsotonII in the holder and select Flush aperture to clean up.
Plating for viable counts
Vortex your sonicated sample again. Use it to make appropriate serial dilutions to plate for viable cell counts. Typically, I plate 100 µl of a 10-4 dilution, made by 4 dilutions of 100 µl culture into 900 µl water or 2 dilutions of 10 µl culture into 990 µl water. Pay attention to your pipetting technique to ensure accurate dilutions. Pipet 100 µl of the final dilution onto a (labeled) plate and spread evenly by your favorite method. I prefer a turntable and sterile 0.2 ml glass pipet. If your colony counts start getting above ~300/plate, you should plate less. I shoot for 100-300 colonies/plate.
Plating for drug resistance
If you are monitoring drug resistances, plate the appropriate volumes of culture on them as well. 250 µl is about the limit you can comfortably plate without puddles forming. If you need to plate more than this, spin down the volume and resuspend the cell pellet in a smaller volume. It is most accurate if you make 1 tube for each plate and plate all of it, rather than spinning a large sample and trying to resuspend it in exactly the correct volume to split up.
Let the plates dry on the bench before transferring them to the 30C room. I keep my plates in a plastic bag to keep them humidified in the warm room.
Sampling for DNA
If I'm sampling for DNA and RNA, I generally do all of the above first, then go back for the other samples. Taking a sample for RNA probably perturbs the culture a bit, so it should be the last thing you do.
For DNA sampling, I use a modified Hoffman and Winston yeast DNA prep. Collect 10 ml of cells from the effluent tube, spin them down, and resuspend in 0.5 ml of the sorbitol buffer. Transfer to a 1.5 ml eppendorf. Freeze –80C.
Sorbitol Solution
|
45 ml |
2 M Sorbitol |
|
10 ml |
1 M Tris pH 8 |
|
20 ml |
0.5 M EDTA |
|
25 ml |
Water |
Sampling for RNA
One RNA microarray requires ~50 µg of total RNA. ~5 ml culture is adequate to ensure this yield, and it is such a small fraction of a 300 ml culture that it should not perturb the chemostat much. First, label a 15 ml Falcon tube for the filtrate and a 2 ml eppendorf tube for the filter. Using a ring stand, set up the small filter apparatus with the stopper assembly. The stopper is just 2 connector fittings jammed into the top of a Falcon tube-sized stopper. One of the fittings is connected by a piece of tubing to the filter and the other goes to the vacuum. Fit the stopper into the 15 ml Falcon tube for the filtrate. Clamp or screw down a 25 mm filter in the filter apparatus, depending on which one you're using. Hook the vacuum tubing to the other stopper port. Rinse the apparatus with milliQ water before using to remove any trace of previous samples. Even a tiny bit of, say, sulfur-limited media contains a huge amount of phosphate relative to the filtrate from a phosphate-limited culture.

Figure 15. Small filtering apparatus.
You also need a bucket of liquid nitrogen. Get another Falcon tube to collect the culture. Loosen the screw holding the drop tube. Put the end of the effluent tubing in the Falcon tube. Plunge the drop tube into the culture. When culture starts pouring from the tube, quickly pull the drop tube out of the culture. You should have about 5 ml of culture in the tube. Turn on the vacuum and dump the culture into the filter assembly. Let it vacuum through. Remove the clamp and glass funnel. Disconnect the vacuum. The order is important to prevent cells from sticking to the glass funnel and to allow all the filtrate to get sucked into the collection tube. Without disturbing the film of cells, remove the filter with a spatula. Fold it and insert into the 2 ml eppendorf tube. Close the tube and put it in the liquid nitrogen. Readjust the drop tube to the appropriate height. Rinse the filter apparatus with milliQ water and repeat for the remaining chemostats. When you've finished collecting all your samples, transfer the tubes to –80C.
Cleanup
Make sure to clean up after yourself once you finish sampling. You want to have all the plating and measurements done shortly after you take the sample, so I generally leave a bit of a mess in my wake and clean it up while my plates dry. Where it's easy to clean up as I go, I do it (i.e., sonicator). In particular, make sure you wash out the Coulter counter cups with DI water and flush the Coulter counter aperture with fresh diluent.
Counting colonies
The different plates will need to grow for different amounts of time. YPD plates need to be left for two days and minimal for 3. Canavanine and 5-FU are good at 4, and alpha-aminoadipate requires 7 days. The most important thing is to be consistent about which day you count the colonies. If you deviate, make sure to write it down. Over the course of an evolution, you may see changes in colony size that require changes in the incubation time. My favorite colony counting technique uses the New Brunswick counter. It contaminates the colonies since you touch them with the stylus, but it's fast and accurate. If you do need to save the plates for some reason, a pen and a clicker work fine too. Sometimes overgrowing the plates can reveal interesting colony morphologies. Record any observations about atypical colony size or morphology.
Media Replacement
You will need to change the media carboy every few days. Plan ahead so your 1X salts can cool overnight before you actually need them. Since we don't have an excess of carboys, please don't stockpile media too much in advance. I think it is best to store the autoclaved 1X salts until you need them, and then add the appropriate additives. Certainly don't prime the supply tubing until right before use. If you have any media leakage, you will get contamination of the tubing. I like to sample before I change the media. I also tend to be a media miser and try to get every last bit from a carboy before changing to a new one. There are a couple of ways to set up the chemostats so that this is easy and riskfree.
If you are running chemostats off of just one carboy, you will have to use this first option. When you are down to the last bit of media, put an eppendorf tube rack under the front of the carboy to tilt it back and pool the remaining media. Adjust the metal drop tube feeding into the chemostat so that it sits at the deepest part of the pool. Check that you didn't break the siphon. You should be able to get almost all the remaining media this way.

Figure 16. Media conservation trick.
The second technique works if you are feeding your chemostats off two media vessels connected by a Y connector. You can replace a media carboy whenever the total media remaining in both carboys will fit into only one. First, drain all the media into one carboy by placing one carboy on a lower shelf than the other. The lower carboy should start to fill up. You can prop up the top carboy as described above to drain the maximum amount of media. Clip the end of the tubing from the empty carboy. Replace the other media carboy on the higher shelf.
With either technique, watch the supply closely. You don't want to forget about it and run out of media. Once you've almost run out of media, or if you are leaving and the media will run out before you get back, replace the carboy. Clamp the end of the tubing, untwist the fitting, and twist it onto the new carboy's fitting. Unclamp the new carboy and watch to make sure the media starts flowing. Write down the time. You may also want to take a sample of the media for analysis. We have had chronic problems with variable media, so it's good to keep track.
See the Filtrate section for advice on nutrient assays.
Data Analysis
Once you've got some sampling data, you'll want to analyze it. At the beginning of a run, it's important to calculate the dilution rate to make sure the pumps are behaving and the settings are correct. The dilution rate is a simple relation of the effluent volume, length of time (in hours) effluent collected, and chemostat volume:
D = effluent volume/(time * chemostat volume)
The dilution rate is in units of hr-1. It is also sometimes called omega.
Since your chemostats will all be running at different rates, either by experimental variation or by design, generations is often a more useful metric than time for graphing things and talking about run length. The chemostat literature talks about two different types of generations: a culture generation, i.e. one volume replacement of the chemostat, and the cell generation, i.e. the doubling the cells must undergo to keep up with the dilution rate. Since some cells get diluted out before they can divide, the culture as a whole must actually double faster than the chemostat volume replacement rate. I usually calculate the cell generations elapsed since I last sampled:
You can cumulatively add up the generations for every sampling point to get a column for making scatter plots.
For measuring drug resistance frequency, I usually add up the total number of colonies on all accurately counted plates and divide by the total volume plated to get resistant cells/ml. Then I divide that number by the coulter count/ml to get a frequency. I find the coulter count is much more well-measured than the viable plate counts, so I use it even though it overestimates the viability.
Harvesting
At the end of the chemostat run, you can harvest the cells to make RNA, DNA, media filtrate, and yield measurements. Make sure you've already done all the sampling you want before you harvest. For example, you may want to take a micrograph.
Setup for harvesting
The key to harvesting cells for RNA is to be as fast as possible. Audrey Gasch famously studied the huge number of genes whose expression changes in the face of almost any stress. This stress response sets in within 5 minutes of perturbation and affects almost 1500 genes. Avoid it at all costs.
Because of the time pressure, make sure everything is completely set up before you start sampling. Using a pen that will stand up to liquid nitrogen and the -80, label 3 15 ml Falcon tubes, 2 for RNA preps and one for filtrate. Label extra tubes for media samples. Get a big bucket of liquid nitrogen. Get the metal tongs if you prefer not to dunk your hand in liquid nitrogen. Find the screwdriver. Get the filtering apparatus together and clean it. Make sure the vacuum is working properly. Get your pipetaid and clean 50 ml pipets. Find your favorite 100 ml graduated cylinder. Untangle a pile of clips. Find your forceps. Get a box of 0.45 micron filters and make sure you have enough to complete the harvests. Make sure there is room in the -80 for all your tubes. Know where you can quickly find a backup pipetaid or extra filters in case you accidently pull culture into the pipet. Know where the backup filtering apparatus is in case you break something.
Weigh a 0.45 micron filter for each yield measurement. Write down the weight and fermenter number on the little piece of paper that separates the filters. Make a foil clamshell for each filter and paper pair so you can easily carry them around and keep them separate from the others.
To set up the filter apparatus, find a 1 L Erlenmeyer flask with a sidearm. There's a heavy one around which is particularly nice. Lubricate the sidearm and the end of the vacuum tubing with some water and attach them. Put the stopper with the filter support in the top of the flask. Make sure the metal support is seated in the glass part correctly. The mesh should be level with the glass rim, not forming a depression. Have the clamp and the funnel nearby.
Harvesting
The active harvesting process should take about 5 minutes. Cleanup and preparation will take another 5 minutes. First, center a 0.45 micron filter on the filter support. Place the funnel on top and carefully clamp it all together. Start the vacuum. Listen for any whistling noises that may indicate a leak in the seal. If you do get whistling, make sure that the filter is centered and free of wrinkles. Some fraction of the filters have cracks or holes that will interfere with the harvest. Replace the filter with a new one if this seems to be the case.
Filter Stopper Vacuum Sidearm flask Clamp Funnel

Figure 17. Large filter apparatus.
Remove the tops from 2 properly labeled 15 ml tubes. Put the tubes in the bucket of liquid nitrogen. Loosen the cap of the third 15 ml tube and place it near the filtering apparatus. Find the correct filter for the yield harvest and place it near the filtering apparatus.
Record the time. With the screwdriver, loosen the screw of the drop tube on the chemostat. Put the effluent tube into a 100 ml graduated cylinder. Push the drop tube down into the middle of the culture. Clip off the media flow immediately. (This avoids delivering excess limiting nutrient to the chemostat while it's being harvested. I have no real evidence that it does anything, but I do it anyway.) Culture should start to eject into the graduated cylinder. As you collect the 100 ml, make sure the drop tube remains submerged in the culture. Once you've collected about 95 ml, pull the drop tube up above the top of the culture. You should have about 100 ml now. Pour the culture into the funnel of the filter apparatus. Watch to make sure it is filtering properly and that no cells are making it into the flask. You can refilter if you have this problem, but try to avoid it.
Once the culture has completely filtered through, remove the clamp and then the funnel. Pull the vacuum hose off the sidearm. This order is important to keep cells from sticking to the funnel. Use forceps or a spatula to lift the edge of the filter, avoiding the cells in the center. Carefully roll up the filter. Dump out the liquid nitrogen in a 15 ml tube, pop the rolled up filter inside, loosely cap it, and dunk it back into the liquid nitrogen. Take the flask and pour filtrate through the sidearm into the room temperature 15 ml tube. Put that tube in the -20.
Put a new filter on the apparatus and reassemble it. Turn the vacuum back on. Collect another 100 ml in the same way as before. Filter it as before and put the filter in the other tube in the liquid nitrogen.
Get the weighed yield filter and again reassemble the filter apparatus. Collect ~60 ml culture from the chemostat. Use a 50 ml pipet to measure out exactly 50 ml of culture. Add this to the funnel. Be particularly careful this time to unclamp, remove funnel, and release vacuum in that order to make sure you collect all of the cells on the filter. Also, be very careful not to scrape any cells off the filter while handing it. Return the filter to its foil clamshell and let air dry. Make sure the cell side of the filter is not sticking to the foil.
Turn off the chemostat by going into the appropriate fermenter menu, hitting 1, toggling to on, and hitting enter.
Wash out the filter apparatus and the graduated cylinder with DI water. Let excess water drain onto a paper towel. Reset for the next harvest.
Once you are finished with all the harvests, you can clean up and make final observations about the look and smell of the cultures. Make sure you move all the RNA harvests from the liquid nitrogen to the -80.
Unhook the air flow and turn it off at the gauge. If you leave the air running, you will unnecessarily drain the backup air if the house air fails. Unplug any probes you had attached. Disconnect the pump tubing from the media. Take a sample of the media if you wish. Once everything is disconnected, remove the chemostat from the housing. Undo the clamp and lift out the metal assembly. Check it for cell growth. The recessed screws on the bottom are a particularly obvious place to look. If you see any cell growths, write it down. Put the cover over the temperature probe plug and set the assembly aside. Swirl the remaining culture. Does it have chunks? Is there wall growth? Write down any observations. Smell the culture. Compare it with any other cultures you harvested. The different limitations all have very distinctive smells. Phosphate and sulfur limitations smell very similar, with a fruity, sweet, sharp smell that has some almond in it. Glucose limitations smell awful, like sweatsocks, and even worse when you have other additives. Some other scents that may be present are bready and acrid. Try to be as descriptive as possible and ask others for their opinions. Write down everything.
You can use the remaining culture for a DNA prep. Spin the cells down and resuspend them in 0.5 ml sorbitol solution (recipe is in the sampling section). Transfer to a 1.5 ml eppendorf tube. Freeze at -80C.
Clean the entire chemostat well with DI water. If you have any obvious wall growth, scrub the walls with a wet paper towel. Rinse well. Let the pieces air dry on a towel somewhere safe and out of the way. Cap the DO probes so they don't dry out.
Clean up any spilled culture or media in the chemostat room. Sometimes a spill will spread under the chemostat. Try to soak it all up and wash the bench with diluted Contrad 70 or bleach. Media spills are a haven for contaminants.
Sample Processing
That's it. You now have a lot of data and a freezer full of glycerol stocks, cell samples for RNA and DNA, and filtrate samples. Next you will want to process them. Once you have RNA or DNA for microarrays, see my microarray protocols at genomics.Princeton.edu/dunham
Filtrates
You may want to measure metabolites and nutrients present in the filtrate. There are a number of commercial kits available from R-Biopharm to measure glucose, ethanol, and many other molecules. Chen, Toribara, and Warner have a great phosphorus assay (see references). It's very easy and accurate, and has a good range. I haven't found a very reliable way to measure sulfur yet, though. You can also measure the pH of the filtrate.
Culture revival
To revive cells from the glycerol stocks for further study, you should streak them to plates. Be careful about the choice of media, though. Some evolved strains no longer grow well on the rich YPD we generally use for day-to-day growth. You may have to make special low-nutrient plates to ensure good growth. You may want to do a test of different media formulations. Scrape out a nice chunk of the glycerol stock and resuspend it in some media. Count a sample in the Coulter Counter to gauge how much to plate. Assume about a 50% revival. Plate an aliquot to each of your types of plates, let grow up, and count colonies. I find pretty striking differences. Phosphate- or sulfur-limited strains seem to do the best on YPD, while some glucose strains like low glucose (0.8%) minimal media better.
No matter what media you choose, watch out for revertant or suppressor colonies that arise on the plates. Always pick an average colony. When growing up culture in liquid media, make only as much cells that you need. This minimizes the number of generations of selection in batch that might allow a revertant/suppressor to take over. Also, don't serial transfer from a culture. Always go back to a fresh colony from the glycerol stock.
DNA prep
Hoffman-Winston prep modified by Maitreya Dunham and Cheryl Christianson.
Before you start, make the lysis buffer and TE+RNase, weigh out the glass beads into individual servings, and label all your tubes. Process only the number of tubes you can fit in your vortexer at one time (for us that's batches of 8). Use phenol-resistant gloves.
Gently thaw cells and spin to pellet. Decant most of the super, leaving just enough to resuspend the pellet completely.
To resuspended pellet, add:
200 ml lysis buffer (recipe below)
200 ml 25:24:1 phenol/chloroform/isoamyl alcohol (alcohol optional)
300 mg acid-washed glass beads (~500 micron size range)
Vortex 8 minutes. We've found this added vortexing increases the yield substantially without obviously shortening the DNA on a 1% gel. We use a small foam multi-tube attachment, ~4 inches in diameter. Do not use one of those funny rack vortexers or a large multitube attachment on a normal vortexer. You should make sure your setup actually vortexes the tubes adequately. If you get low yields, this is a key step to check.
Touch spin in a low speed minifuge to get the phenol off the lid.
Add 200 ml TE. Invert to mix.
Spin 5 min max speed in a microcentrifuge.
Carefully transfer aqueous (top) layer to a new tube without catching interphase junk.
Add 1 ml room temp 100% ethanol. Invert to mix.
Spin 2 min max speed.
Decant super and resuspend pellet in 400 ml TE+30 mg RNaseA. The pellet may not resuspend easily. As the incubation proceeds, you can usually get the whole thing to resuspend, though.
Incubate 30 minutes at 37C. We've lengthened this digestion from the original 5 min to reduce RNA contamination and to make sure the entire pellet gets into solution.
Add 10 ml 4 M ammonium acetate and 1 ml room temp 100% ethanol.
Invert to mix.
Spin 2 min max speed.
Decant super completely and dry pellet. We leave the tube inverted on a kimwipe on the bench for about 10 min.
Resuspend in 50 ml TE.
Measure DNA concentration using a fluorometer or other DNA-specific method (i.e., NOT the spectrophotometer). Even with the RNase treatment and ammonium acetate precipitation, there's a lot of RNA contamination in these preps.
Total yield should be 10-20 mg. DNA should restriction digest easily.
Lysis buffer for DNA
We make this fresh each time for some unknown reason.
2% Triton X-100
1% SDS
100 mM NaCl
10 mM Tris pH 8
1 mM EDTA
RNA prep
Hybrid of the DeRisi protocol (www.microarrays.org) and a standard acid-phenol prep circulating around the Brown/Botstein labs circa 2001
The protocols for the large and small preps are more or less the same with minor volume and centrifuge differences. The phase lock gel makes everything so much easier, but if you aren't using it, add in 1 or 2 extra chloroform extractions to clean things up.
Use RNase free reagents and glass-/plastic-ware throughout! Remember to use glass pipets with chloroform.
Lysis buffer for RNA
(100 ml)
|
2 ml |
0.5 M EDTA |
|
5 ml |
10% SDS |
|
1 ml |
1 M Tris pH 7.5 |
|
92 ml |
RNase-free water |
RNA prep for 5 ml daily samples
Remove a manageable set of samples from the –80. They should be in 2 ml eppendorf tubes.
Before they thaw, add 750 µl lysis buffer. Vortex, trying to get all the cells off the membrane.
Add 750 µl acid phenol. Vortex.
Incubate 1 hour 65C, vortexing every 20 minutes.
Fish out the filter and discard.
Ice 10 min.
While they are incubating, spin the 2 ml heavy phase lock gel (PLG) tubes for 30 sec full speed in a room temperature microcentrifuge. Set aside.
Spin lysate 5 min.
With a pipet, transfer the top aqueous layer to the PLG tube.
Add 750 µl chloroform. Invert to mix. Do not vortex!
Spin 5 min.
Pour aqueous layer into a new 15 ml Falcon tube.
Add 75 µl (or 1/10 volume if you lost some) 3 M sodium acetate. Mix.
Add 1.5 ml (or 2 volumes) ethanol. Mix.
Incubate –20C >30 min (preferably overnight).
Spin 3000 rpm 10 min.
Wash pellet 2X with 70% ethanol, with 2 min 3000 rpm spins between washes.
Air dry inverted on the bench 30 min.
Dissolve pellet in 25 µl water. You can speed up the dissolution by heating the sample or by pipetting the pellet up and down, but I prefer a gentler room-temperature-with-frequent-flicking approach.
Measure the concentration with the spectrophotometer. You should get about 50-60 ug, just enough for an array. If using the nanodrop, use the RNA undiluted.
Check the quality of the RNA on the Bioanalyzer or a gel. You will probably have to dilute it ~1/10 to run on the Bioanalyzer.
RNA prep for 100 ml harvest
Remove a manageable set of samples from the –80. They should be in 15 ml Falcon tubes.
Before they thaw, add 4 ml lysis buffer. Vortex, trying to get all the cells off the membrane.
Add 4 ml acid phenol. Vortex.
Incubate 1 hour 65C, vortexing every 20 minutes.
Fish out the filter and discard if you wish. I generally don't for the larger samples, but the filter can sometimes interfere with the phase separation.
Ice 10 min.
While they are incubating, spin the 15 ml heavy phase lock gel (PLG) tubes for 2 min 1500xg in a room temperature swinging bucket centrifuge. Set aside.
Spin lysate 10 min 3000 rpm.
With a pipet, transfer the top aqueous layer to the PLG tube.
Add 4 ml chloroform. Invert to mix. Do not vortex!
Spin 5 min 1500xg.
Add another 4 ml chloroform to the same tube, invert to mix, and spin again.
Pour aqueous layer into a new 15 ml Falcon tube.
Add 400 µl (or 1/10 volume if you lost some) 3 M sodium acetate. Mix.
Add 8 ml (or 2 volumes) ethanol. Mix.
Incubate –20C >30 min (preferably overnight).
Spin 3000 rpm 10 min.
Wash pellet 2X with 70% ethanol, with 2 min 3000 rpm spins between washes.
Air dry inverted on the bench 30 min.
Dissolve pellet in ~250 µl water, adding more if necessary. You can speed up the dissolution by heating the sample or by pipetting the pellet up and down, but I prefer a gentler room-temperature-with-frequent-flicking approach.
Measure the concentration with the spectrophotometer. You should get several mg. If using the nanodrop, you will almost certainly have to dilute the RNA. 1/2 is a good guess, or dilute 1/20 and use the same sample for the Bioanalyzer. You could also run a gel to check the quality.
Problems
|
Problem |
Explanation and Solution |
|
|
|
|
Pump squeaking or skipping |
|
|
|
|
|
Flow rate wrong |
Pump problems as above. Program wrong, check flow settings. Effluent tube not in effluent vessel (should be accompanied by spill). Kink in tubing. |
|
|
|
|
Vessel fills up, won't eject effluent |
Air leak. Check clips on tubing, o-ring seating, and rubber bands on bubblers |
|
|
|
|
Additives won't vacuum into carboy |
Air escape. Check that clips are secure and that the stopper is on. Reposition drop tubes straight up-and-down. Don't leave drop tubes angled in the stopper. May need to replace stopper if too stretched. |
|
|
|
|
Pink colonies on drug plates |
Common contaminant. Change effluent tubing before sampling. If that doesn't help, see advice below on other contaminants. |
|
|
|
|
Contamination on viable count plates |
Either contamination of the vessel or the water/media used to dilute the cells. Check vessel culture by directly sampling through one of the top ports. Replace water/media. |
|
|
|
|
Washout |
Dilution rate set too high. Check settings. Media missing an ingredient. Media carboy contaminated. |
|
|
|
|
Mystery spill |
Either water or media. Use the glucose test strips to decide. If media, check for loose tubing connections or holes in the tubing. If water, check the water tubing and faucet for leaks. |
|
|
|
|
Neighboring chemostats correlated |
Media inconsistency. Measure limiting nutrient concentration of each carboy of media. Could also be a coincidence. |
|
|
|
|
Media flow stops |
Broken siphon. Prime tubing before connecting carboy. Check for air bubbles and clear them by blowing into the big air filter on the carboy. Check for holes in the tubing. Kinked tubing |
|
|
|
|
Pumps all come on, screen wacky. |
Corrupted memory card. Swap the card with another one and see if that solves the problem. (Port is on top section of the chemostat, on the left side. Turn it off before servicing.) Can also try reinitializing card by hitting escape as the machine turns on, then enter to initialize. |
|
|
|
|
Low carboy volume after autoclaving |
Autoclave malfunction. Loose clip on tubing. |
|
|
|
|
Cloudy filtrate during harvest |
Faulty filter. Replace and refilter. Filter not centered on apparatus. Reposition and refilter. |
|
|
|
|
Funny noises, strange behavior |
Sharpie stuck inside the machine. |
|
|
|
|
Unique problem with machine |
Call Andy at ATR 1-800-827-5931 |
|
|
|
Chemostat References
Chen, P.S., T.Y.
Toribara, and H. Warner. 1956. Microdetermination of Phosphorus. Analytical
Chemistry 28: 1756-1758.
Dykhuizen, D.E. and
D.L. Hartl. 1983. Selection in chemostats. Microbiological Reviews 47: 150-168.
Dykhuizen, D.E. 1993.
Chemostats used for studying natural selection and adaptive evolution. Methods
in Enzymology 224: 613-631.
Ferea, T.L., D.
Botstein, P.O. Brown, and R.F. Rosenzweig. 1999. Systematic changes in gene
expression patterns following adaptive evolution in yeast. Proceedings of the
National Academy of Sciences of the United States of America 96: 9721-9726.
Kubitschek, H.E. 1970.
Introduction to Research with Continuous Cultures. Prentice-Hall, Inc.,
Englewood Cliffs, NJ.
Paquin, C. and J.
Adams. 1983. Frequency of fixation of adaptive mutations is higher in evolving
diploid than haploid yeast populations. Nature 302: 495-500.
van Dijken, J.P., J.
Bauer, L. Brambilla, P. Duboc, J.M. Francois, C. Gancedo, M.L.F. Giuseppin,
J.J. Heijnen, M. Hoare, H.C. Lange, E.A. Madden, P. Niederberger, J. Nielsen,
J.L. Parrou, T. Petit, D. Porro, M. Reuss, N. van Riel, M. Rizzi, H.Y.
Steensma, C.T. Verrips, J. Vindelov, and J.T. Pronk. 2000. An interlaboratory
comparison of physiological and genetics properties of four Saccharomyces cerevisiae
strains. Enzyme and Microbial Technology 26: 706-714.
Supplies and Suppliers
ATR
www.atrbiotech.com
Andrew Magno
Sixfors 500 ml Mammalian Cell Culture System
and accessories
Fisher
www.fishersci.com
Nalgene 115 ml 0.45 micron filter unit
(for filtering in additives if making media by autoclaving)
121-0045
12 pack
$46.21
Nalgene fast PES bottle-top filter, 90 mm diameter, 0.2 micron
(for filtering whole carboy of media)
597-4520
12 pack
$124.65
Bottle, gas, 250ml
(air bubblers)
03-038-B
$102.35
Clamp, pinchcock (12/pk)
05-867
$17.14
Tubing 3/8x9/16 (50ft/cs)
(braided heavy duty tubing)
$74.33
Tygon sanitary silicone tubing,
formula 3350 (50 feet)
(large bore tubing)
02-587-1Q
$131.20
Tygon tubing 3/32 ID 7/32 OD, formula 3350 (50 feet)
(small bore tubing)
02-587-1E
Fisherbrand polypropylene clamp
(small tubing clips)
Pack of 12
05-869
$6.25
Millipore FG50 0.2 micron filters
(for venting on media carboys)
10 pack
SLFG 850 10
$83
Kontes Ultra-ware 47 mm microfiltration assembly with stainless steel support
K953805-0000
$127
Fisherbrand
Glass Microanalysis Vacuum Filter Holder
25
mm, stainless steel support
09-753G
$167.16
New Brunswick Scientific BioFlo 2000 Medium Reservoir Assembly
(carboy with hardware)
14-282-39
$758
13L pyrex carboy, case of 4
(carboy without hardware)
02-887-3
$603.92
Fisherbrand Solid Neoprene Rubber Stoppers. size 12, 5/pk
(for home-made carboy hardware)
14-141R
GE Osmonics
www.osmolabstore.com
MAGNA filters, nylon, supported,
0.45 micron, 47mm (100/pk)
R04SP04700
$90
MAGNA filters, nylon,
supported, 0.45 micron, 25mm (100/pk)
R04SP02500
$68
Ace Glass
Glass chemostat vessel
Special order via Paul Benn
(Tell him Maitreya from Princeton sent you.)
1-800-223-4524
$185 @ for 6 or $165 @ for 12
Masterflex/Cole Parmer
www.masterflex.com
Female luer PP fittings;
1/8" ID tube
package of 25
HV-06359-37
$9.25
package of 10
HV-30703-92
$16.25
Polypropylene fittings; male
slip x 1/8" hose barb
package of 25
HV-06359-17
$9.25
Plastic luer fittings, Male
luer lock x 1/8" hose barb
package of 25
HV-30504-10
$9.50
Pump tubing, various
Small filters for gas
filtration
box of 100
HV-02915-22
$125.00
R-Biopharm AG
www.r-biopharm.com
Ethanol test kit
10176290035
$82
D-Glucose test kit
McMaster-Carr
www.mcmaster.com
Ultra-Precision Extension
Springs
(springs for bubblers)
$8.57